

1. Введение
Аритмогенная кардиомиопатия (AК) — это заболевание сердечной мышцы, клинически характеризующееся опасными для жизни желудочковыми аритмиями (ЖА) и стремительно прогрессирующей дистрофией желудочкового миокарда с фиброзно-жировой заменой [1].
Говоря подробнее, АК обычно характеризуется систолической дисфункцией правого желудочка (ПЖ), структурной деформацией ПЖ, формированием злокачественной желудочковой тахикардии (ЖТ) с развитием высокого риска внезапной сердечной смерти (ВСС) [2]. У спортсменов, особенно тех, которые занимаются беговыми видами спорта с упором на выносливость [3], а также у людей в возрасте около 35 лет [4] АК является достаточно частой причиной ВСС.
Предполагается, что патология имеет распространенность 1: 2000-1: 5000, таким образом, можно сделать вывод, что AК относится к числу редких заболеваний [5]. Фактическая распространенность может быть и больше, поскольку диагноз может быть пропущен клинически и при посмертном вскрытии. Кроме того, на ранних стадиях AК более напоминает ионные каналопатии, чем другие неишемические кардиомиопатии.
Наибольшую известность получило название аритмогенная кардиомиопатия правого желудочка (АКПЖ), которое было зарегистрировано вследствие неопределенного количества сведений о наиболее частом поражении в данной локализации [6].
Однако недавно было признано, что деструкция наблюдается не только в правом сердце: отмечались частые случаи повреждения либо левого, либо обоих желудочков, что привело к принятию более широкого термина «аритмогенная кардиомиопатия» [7].
АК является слабо изученным заболеванием, но достоверен тот факт, что оно оказывает существенное влияние на развитие и выраженность феномена внезапной сердечной смерти. Аритмогенная кардиомиопатия чаще имеет очень характерную патологоанатомическую картину, но именно патофизиологические основы до сих пор не определены. Множество теорий предполагает множество различных терапевтических подходов, направленных на ту или иную нозологию, что, в свою очередь, вносит разрозненность во мнениях лечения у разных клиницистов. Является ли данная патология кардиохирургической, кардиологической, генетической, либо совершенной другой направленности?
В нашем обзоре представлены наиболее современные гипотезы, касающиеся патогенеза АК, с разных точек зрения, а также описаны особенности диагностики и лечения.
2. Патологоанатомическая картина
По мере развития болезни миокард желудочка проявляет прогрессирующую дегенерацию кардиомиоцитов с заменой на жировую и фиброзную ткань. Характер и распределение дегенерации миокарда весьма характерны и отчетливо отличаются от таковых при других кардиомиопатиях [8]. AК характеризуется прогрессирующей фиброзной заменой желудочкового миокарда, включая ПЖ и ЛЖ, с относительной сохранностью перегородки.
Сегменты свободной стенки ПЖ, которые испытывают наибольший механический стресс во время сердечного цикла - задний сегмент ниже трикуспидального кольца, вершина и отводящий тракт ПЖ - претерпевают наибольшие изменения. В общем, эндокардиальные трабекулярные мышцы ПЖ и межжелудочковая перегородка остаются, как правило, не вовлечены. Когда задействован левый желудочек, дегенерация миокарда и фиброз наиболее заметны в субэпикарде и среднем миокарде латеральной стенки. Это объясняет морфофункциональные аномалии стенки желудочка, которые могут отсутствовать на ранних стадиях.
Атрофия миокарда происходит постепенно со временем, начинается с эпикарда и в конечном итоге распространяется трансмурально. Эту сущность не следует смешивать с болезнью Ула, врожденным пороком сердца, при котором миокард ПЖ не развивается во время эмбриональной жизни [9].
Гистологическое исследование выявляет островки выживших миоцитов, чередующихся с волокнистой и жировой тканью [10, 11] (Рисунок). Первоначально были предложены две патологические формы заболевания: жировая и фиброзно-жировая [11]. Однако теперь признано, что значительный рост эпикардиальных адипоцитов может возникать в ПЖ у лиц пожилого возраста или людей с ожирением, которые в крайних случаях могут распространяться фокально к поверхности эндокарда и, по крайней мере, поверхностно напоминают картину, наблюдаемую в AК. Поэтому инфильтрация жировой ткани без воспаления и дегенерации миоцитов больше не считается достоверным критерием диагностики AК [12, 13], но при сочетании с фиброзом замещающего типа и дегенеративными изменениями миоцитов можно достаточно правильно установить данную патологию.
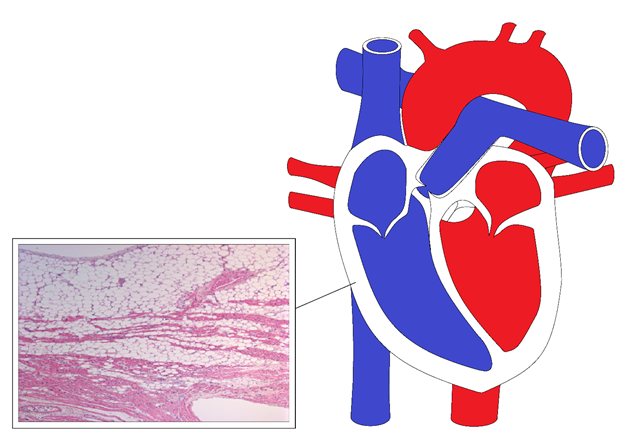
Рис. Распределение островков выживших миоцитов, чередующихся с волокнистой и жировой тканью в миокарде правого желудочка средца.
Некротические изменения миоцитов нередко могут быть связаны с воспалительными инфильтратами [14]. Сообщалось о воспалении миокарда до 75% сердец при аутопсии. Кроме того, есть гипотеза, указывающая на мононуклеарное воспаление, преимущественно лимфоцитарное, которое может быть обширным и связанным с ним с областями дегенерации миоцитов, создавая картину, подобную той, которая наблюдается при вирусном миокардите. Роль воспаления в AК остается в значительной степени неопределенной. [15]
Обнаружение вирусных геномов позволяет продвигать инфекционную вирусную этиологию, но, скорее всего, либо вирусы являются невиновными наблюдателями, либо дегенерация миокардиальных клеток может служить средой, благоприятствующей вирусному поселению [16]. Также доказан апоптотический механизм смерти миоцитов [17,18].
3. Патофизиологическая картина
Патологоанатомическая картина АК во многом определена и описана достаточно подробно, чтобы с большей долей вероятности при вскрытии предположить наличие патологии. Нерешенные проблемы данной патологии заключаются именно в патофизиологической и этиологической принадлежностях. Происхождение AК до сих пор в значительной степени неизвестно, но различные теории, пытаются обосновать то или иное проявление болезни. Первые исследования [Fontaine et al., 1977] указывали на наличие врожденного дефекта из-за аномалии эмбрионального развития ПЖ, чем и объясняется первичное название болезни Аритмогенная дисплазия правого желудочка. Как указана выше, патология находила сходство с болезнью Ула, но фиброадипозное замещение миокарда отсутствует в данном случае [Pamuru et al., 2010], что также указывает на некорректность термина «дисплазия» и объясняет причину замены названия на «кардиомиопатию» [Basso et al., 2010].
Мы считаем, что описание возможных патогенетических форм АК стоит начинать от объяснения генных перестроек, влияющих на клеточные взаимодействия. В настоящее время это наиболее целесообразный подход к данному заболеванию в связи с наличием огромного количества сведений о семейной предрасположенности АК. Таким образом, стоит демонстрировать современные понятия о патологии со стороны теорий, приведенных в классификации ниже:
Варианты первичных причин АК:
-Теория генных мутаций;
-Теория аномальной внутриклеточной сигнализации;
-Теории трансдифференциации и дистрофии кардиомиоцитов;
-Теория аномальной клеточной адгезии по типу «клетка-клетка»;
-Теория общего ремоделирования сердечной мышцы;
Варианты вторичных причин АК, усугубляющих течение заболевания:
-Воспалительная теория;
-Теория внезапного изменения гемодинамического объёма крови.
4. Варианты первичных причин АК
Варианты первичных АК – это в первую очередь генные перестройки, как было указано выше. Наибольшее внимание привлекают именно данные теории, поскольку изменения в одном гене могут приводить к клинике, напоминающей АК. Безусловно, симптоматика АК, как будет описано на примере клинического случая, пока мало описана и во многом вариативна, поэтому генная кардиомиопатия с угрожающими жизни аритмиями возможно либо имеет множество фенотипических групп, либо содержит в себе патологию иного уровня.
4.1. Теория генных мутаци
АК в настоящее время преимущественно относят к генетическим заболеваниям. Считается, что примерно в 60% случаев АКПЖ-мутация является основной причиной заболевания [19]. Мутации в генах, кодирующих десмосомные белки плакофиллин-2 (PKP2), плакоглобин (название JUP), десмоплакин (DSP), десмоколлин-2 (DSC2) и десмоглин-2 (DSG2), связаны с развитием АКПЖ [20]. Эти мутации преимущественно обнаруживаются в гене PKP-2, но также были замечены в генах, кодирующих не десмосомные белки, такие как рианодиновый рецептор-2 (RYR2), трансформирующий фактор роста-β3 (TGFβ3), ламин A / C (LMNA), трансмембранный белок-43 (TMEM43), десмин (DES), титин (TTN), αT-catenin (CTNNA3) и PLN [20].
Первый ген аутосомно-доминантной АКПЖ был идентифицирован ещё в исследованиях 2000-ых годов, который кодирует RYR2 на хромосоме 1q42-43 [21]. За последние десятилетия в нескольких исследованиях было выявлено более 100 генов и мутаций, участвующих в различных формах сердечных заболеваний, таких как гипертрофическая, дилатационная и аритмогенная кардиомиопатия (ГП, ДК и AК соответственно) [22]. Эти гены в основном кодируют белки сократительной функции, но также и белки, которые участвуют в гомеостазе кальция, и те, которые находятся в интеркалированном диске (ИД), но их патогенность по-прежнему вызывает споры.
Мутации в этих генах оказывают патогенное действие на несколько сердечных клеточных функций, таких как генерация сократительной силы, регулирование силы, трансдукция сигнала, возбудимость и внутриклеточные влияния кальция. С момента открытия первых мутаций, ответственных за развитие АК, внимание к заболеванию, как к наследственной генетической патологии значительно возросло. В первую очередь, так как АК часто приводит к ВСС, данные гипотезы стали приемлемым объяснением неизвестной этиологической принадлежности заболевания, которые в достаточной степени могли бы указать на хотя бы какое-то объяснение патогенеза и патоморфологии, описанных выше.
Интересное клиническое наблюдение продемонстрировали Jeremy W. Docekal and Joseph C. Lee (2017), в котором был описан случай новой генной мутации в DSP. Напомним, что ген DSP кодирует белок desmoplakin, который является важным компонентом десмосом, специализированных белков, которые образуют межклеточные соединения. Мутации в десмосомных генах могут либо нарушать межклеточной адгезию, либо промежуточную филаментную функцию [23].
Пациент - 54-летний мужчина, активно занимающийся спортом. Пробежки на дистанцию 10-12 миль в день не выявляли какие-либо функциональные ограничения. В июле 2013 года потерял сознание во время бега из-за внезапной остановки сердца. Но интересен факт спонтанного возвращения кровообращения с дальнейшим полным восстановлением сердечной деятельности. Пациент отмечал 2 предыдущих случая, когда его сердце внезапно начинало аритмично быстро сокращаться во время занятий спортом, но эти проявления не были достаточно исследованы. Фракция выброса ЛЖ на тот момент была примерно 25%, а МРТ демонстрировало множественные рубцовые области, включающие ЛЖ, значительную дилатацию ЛЖ, но нормальный ПЖ (размер и функция). Диагноз (неишемическая кардиомиопатия) после коронарной ангиографии подтвердил отсутствие доказательств в сторону заболеваний коронарных артерий сердца [24].
В данном клиническом случае демонстрировался вариант АК, имеющий непропорциональную степень дилатации и дисфункции ЛЖ, в сочетании с нормальным ПЖ. Анализ семейной предрасположенности продемонстрировал, что из 5 членов семьи, идентифицированных, как имеющих c.3735_3741dupAAATCGA мутации, 3 имеют объективные доказательства ЛЖ-дисфункции, в то время как 2 - не имеют предрасположенности. У данного пациента же отмечался редкий генетический вариант АК, приводящий к преждевременному синтезу стоп-кодона в экзоне 23 гена DSP. Была проведена оценка состояния пациента с генотипом-положительным АК: ЭхоКГ, Холтеровское мониторирование, ЭКГ, кардиобеговая дорожка. Аномалий не было выявлено.
Конечно же, имеются ограниченные данные, которые сообщают о бессимптомных носителях АК [25], но этот факт лишний раз подтверждает, что этиологическая и патогенетическая принадлежность данного заболевания до сих пор не объяснена в полной мере.
4.2. Теория аномальной внутриклеточной сигнализации
Изучая типичное проявление АК – адипогенез, исследователи [Garcia-Gras E, et al. 2006] выявили особенную роль изменения внутриклеточной сигнализации, проявляющуюся ингибированием пути Wnt. Особенность этого механизма, в первую очередь, объясняется аномальным распределением белков ИД.
Мышам с дефицитом DSP авторы ингибировали путь Wnt / β-catenin / Tcf / Lef, который, как известно, является регулятором адипогенеза, фиброгенеза и апоптоза. Нокдаун DSP в клетках HL-1 вызывал транслокацию JUP в ядро, где он начал препятствовать транскрипционной активности β-catenin / TCF и приводить к адипогенному действию. Кроме того, у мышей, сверхэкспрессирующих JUP, подавление канонического сигнального пути Wnt и индукция экспрессии проадипогенных генов из-за ядерной транслокации JUP приводили к адипогенезу в клетках-предшественниках кардиомиоцитов через C-KIT [26].
Также активно, как и путь Wnt изучается передача сигналов через Hippo / YAP (в ядре YAP взаимодействует с β-catenin для управления экспрессией гена, связанного с Wnt), который тоже, вероятно, связан с АК. Авторы [Chen at al. 2014] продемонстрировали аномальную активацию киназного каскада Hippo, приводящую к фосфорилированию и удержанию YAP в цитоплазме клеток, у которых блокирован PKP2 для имитации АК. В результате была вызвана секреция β-катенина и JUP в цитоплазме с последующим подавлением канонической передачи сигнала Wnt, приводящей к усиленной гибели миоцитов и фиброадипогенезу.
Таким образом, клетки, имеющие отличающиеся от нормальных кардиомиоцитов внутриклеточные сигналы, вероятнее всего, оказывают существенное влияние на адипогенез и фиброгенез: процессы, способствующие деструкции миокарда при АК [27].
4.3. Теория трансдифференциации и дистрофии кардиомиоцитов
Для объяснения патофизиологии АК была выдвинута гипотеза о трансдифференцировке кардиомиоцитов [d'Amati et al., 2000]. Исследователи указывают на генетически детерминированную дистрофию кардиомиоцитов с дальнейшим преобразованием в скопления фиброзно-жировых отложений. Следуя данной гипотезе, d’ Amati и др. поясняют, что кардиомиоциты при АК могут перепрограммироваться и дифференцироваться в адипоциты из-за аномалии генетического кода. Однако в настоящее время, гипотезу оспаривают в следствии ограниченного количества доказательств трансдифференцировочных возможностей взрослых кардиомиоцитов при АК [28].
Дистрофическая теория происхождения AК во многом напоминает о трансдифференцировочном механизме, однако имеет некоторые отличия. В настоящее время данная гипотеза шире распространена и является более одобренной из-за наличия сходств AК со скелетными мышечными дистрофиями [Basso et al., 1996]. Согласно этой гипотезе, фиброзно-жировые отложения в миокарде AК описываются как рубцовая ткань, которая заменяет погибшие кардиомиоциты. Следовательно, генные мутации при AК вызывают не только гибель мышечных клеток сердца и потерю ткани миокарда, но и сигнал об аберрантном восстановлении мышечной массы [Basso et al., 2012].
4.4. Теория аномальной клеточной адгезии
Десмосомы опосредуют клеточную адгезию. Еще до первого описания данного механизма при AК были проведены исследования методом электронной микроскопии, которые продемонстрировали необъяснимое изменения ИД [Guiraudon CM et al. 1989]. Тогда был впервые поднят вопрос об отклоняющейся от нормы адгезии по типу «клетка-клетка» в патогенезе АК.
В недавнем исследовании Sato et al., проводилась работа по изучения влияния PKP2 на АК. Используя монослои неонатальных миоцитов желудочков сердца крыс, в которых PKP2 был ингибирован, исследователи продемонстрировали снижение функции клеточной адгезии [29]. Однако при экспрессии мутантных форм PKP2 или JUP клетки проявляли аномальный ответ и демонстрировали сохраненную межклеточную адгезию, тем самым ставя под сомнение первичную роль адгезии «клетка-клетка» в патогенезе AК [30]. В другом исследовании Asimaki et al. [31] демонстрировали, что уменьшенный контактный сигнал для JUP, по-видимому, является признаком заболевания миокарда у пациентов с AК, что указывает на его возможную роль во внутриклеточной передаче сигналов, а не в процессах адгезии, как это было предложено другими группами [32, 33].
В настоящее время аномальная клеточная адгезия является одной из возможных патогенетических теорий, оказывающих влияние на развитие АК, и она объясняет некоторые аспекты заболевания, но не всю картину. Кроме того, в данном механизме снова на первый план выносится гипотеза генных перестроек, вероятно, напрямую связанная с влиянием на миокардиальные клетки и межклеточные взаимодействия в целом.
4.5. Теория общего ремоделирования сердечной мышцы
Говоря о патофизиологии процессов следует в первую очередь описать те самые аритмии, которые приводят к смертельным исходам. Напомним, что выделяют четыре фазы классического варианта течения АК:
1) «скрытая»: незначительные структурные изменения ПЖ, с или без ЖА;
2) «проявляющиеся нарушения ритма» с симптоматическими угрожающими жизни ЖА, связанными с явными морфофункциональными аномалиями RV;
3) «отказ ПЖ» из-за прогрессирования и распространения заболевания ПЖ;
4) «бивентрикулярный отказ», одновременное поражение ЛЖ [34].
Возвращаясь к аритмиям, стоит отметить, что часто им способствуют электрическое и структурное ремоделирование, из-за чего нарушается связь клеток, их возбудимость и страдает насосная функция сердца в целом [35].
Электрическое ремоделирование заключается в аномалии регуляции ионных и щелевых контактов с расширением внеклеточной матрицы (образование фиброза), что вызывает нарушение передачи импульса между кардиомиоцитами и фибробластами [35,36]. Происходит патологическое увеличение внутриклеточной концентрации кальция из-за дисфункции регуляторных белков, что приводит к дискоординации систолы и диастолы [Qu et al., 2015].
Структурное ремоделирования заключается в гибели кардиомиоцитов, гипертрофии клеток и отложении коллагена (фиброз). В результате происходят те же процессы нарушения распространения импульса между миоцитами. [35]
Рассматривая аритмогенную кардиомиопатию правого желудочка, как наиболее частую форму АК, можно наблюдать классическое проявление структурного ремоделирования, но в сочетании с электрическим: с явными проявлениями ЖА. Как защитный механизм для противостояния сильным и частым механическим воздействиям, отдельные кардиомиоциты плотно соединяются друг с другом через субклеточный домен, имеющий название ИД. ИД интересны с той точки зрения, что они не только физически соединяют кардиомиоциты с помощью десмосом и связных контактов, но также образуют пары кардиомиоцитов для обеспечения быстрого распространения электрических сигналов через щелевые соединения [37]. Накопление экспериментальных данных подтверждает идею о том, что ИД следует рассматривать как сингулярно организованную функциональную сущность, в которой макромолекулярные комплексы механически и электрически взаимодействуют синхронно [38].
Десмосомы, щелевые соединения и натриевые каналы действуют как функциональная триада, в которой изменения в составе одного компонента могут влиять на функцию и целостность других. Ключевым компонентом связи данных комплексов внутри ИД является ANK3 (Ankyrin-G): он объединяет натриевые каналы, щелевые контакты и сердечную десмосому [39]. Межбелковые взаимодействия внутри ИД и те, которые составляют щелевые соединения и натриевые каналы, оказывают существенное влияние на патофизиологию аритмий, особенно, в случае AК. Исследования последнего десятилетия свидетельствует о том, что дестабилизация ИД (через нарушение межбелковых взаимодействий) и отклонения в процессах транспорта его композиционных белков вызывают ремоделирование сердца [40].
В целом, можно сказать, что ремоделирование сердца относится к генетическим аномалиям, поскольку известно, что влияние на возбуждение сердечной мышцы оказывает PLN (phospholamban). Данный ген регулирует потоки ионов кальция, которые активируются при обнаружении потенциала действия из соседних клеток. Увеличение амплитуды ионов натрия пролонгирует этот потенциал действия, что увеличивает трансмуральную дисперсию реполяризации и вызывает аритмогенез. Однако, в экспериментальных моделях AК [41, 42, 43] было обнаружено, что ток ионов натрия был снижен. Эти данные привели к гипотезе о том, что угрожающие жизни ЖА могут возникать у пациентов с АК даже до структурных отклонений из-за электрического разобщения и снижения тока натрия. Несмотря на эти результаты исследований, данную гипотезу еще предстоит доказать у пациентов с AК у человека [43].
5. Вторичные варианты причин АК
Данная немногочисленная группа вариантов причин, усугубляющих АК, вероятнее всего, содержит большее количество гипотез, однако на современном этапе развития учения о кардиомиопатиях выявляет в основном данные вторичные патологии.
5.1. Воспалительная теория
Как было указано выше, исследователи давно предполагают воспалительную теорию АК [16]. Роль инфекционных агентов в спорадических случаях АКПЖ (указано устаревшее название, в связи с давностью исследования) была предложена из-за частого обнаружения воспалительных инфильтратов в миокарде, что свидетельствует о том, что заболевание является следствием миокардита (например, дилатационная кардиомиопатия). Однако, до сих пор не известно воспаление происходит в результате смерти кардиомиоцитов или первично: как иммунный ответ на инфекционное поражение.
Описан случай обнаружения кардиотропного вируса при АК [Bowles et al. 2002]. Энтеровирусные последовательности были обнаружены у семи пациентов, а аденовирусные (тип 5) - у двух других пациентов. Это указывает на связь между АК и наличием вирусного генома (энтеровируса или аденовируса) в миокарде, а, следовательно, объясняет воспалительную теорию. Сходство между АКПЖ и миокардитом (дилатационная кардиомиопатия) с точки зрения гистопатологических данных и дилатации желудочков может объединять этиологическую принадлежность этих вирусов. Несмотря на это, отчетливые патологоанатомические различия между АКПЖ и миокардитом (дилатационная кардиомиопатия) в связи с наличием жировых инфильтратов, наиболее частой локализации в ПЖ и более частых ЖА, свидетельствуют о том, что у пациентов с АКПЖ совершенно другая патофизиология. Она, в свою очередь, может приводить к повышению восприимчивости к инфицированию кардиотропными вирусами. Но является ли воспалительная теория самостоятельной/первичной причиной АК, в настоящее время не определено [44].
5.2. Теория внезапного изменения гемодинамического объёма крови
С точки зрения патофизиологии интересно влияние гемодинамики на больных, страдающих АК (имеющих предрасположенность к заболеванию). Соответствующий патогенез скорее вторичен и лишь усугубляет само заболевание, но в настоящее время, точных сведений об этом нет.
Данная проблема чаще встречается у спортсменов при тех или иных упражнениях на выносливость или требующих быстрого старта. Происходящее изменение объёма крови приводит к устойчивому увеличению постнагрузки на ПЖ и, как следствие, влиянию на инфильтрацию миокарда фиброзно-жировыми клетками [45]. Исходя из этого, закономерно, что постоянные усиленные тренировки на выносливость и прочие тяжелые упражнения ускоряют прогрессирование АКПЖ (в данном случае) у пациентов, предрасположенных к АК.
6. Заключение
В настоящее время вопрос о патогенности мутаций при сердечных заболеваниях и, в частности, при АК является спорным. Анализ многих групп генов идентифицирует большое количество условно патогенных последовательностей с неопределенным клиническим значением. Учитывая, что распространенность мутаций еще не определена, отрицательный генетический тест не исключает генетическую предрасположенность к АК, так как есть случаи бессимптомного носительства. С другой стороны, эти сведения указывают и на условное отношение АК к генетическим мутациям кардиотропного влияния. В связи с этим в настоящее время исследуются и другие патогенетические механизмы, которые указывают на соответствующие этиологические факторы агрессии в сторону сердца.
Важность определения этиологической принадлежности АК очень важна, поскольку до сих пор нет единого золотого стандарта диагностики заболевания. Необходимы выявлять множество критериев, объединяющих различные источники диагностической информации, такие как морфофункциональные аномалии (ЭхоКГ и / или ангиография и / или сердечный магнитный резонанс), гистопатологические аномалии (при эндомиокардиальной биопсии), выявление аритмий (Холтеровское мониторирование, ЭКГ) и изучение семейного анамнеза, включающего генетику.
Положительным фактором, играющим важную роль в профилактике ВСС, является отнесение людей с умеренной или тяжелой дисфункцией ЛЖ к пациентам, подверженным риску АК. Следовательно, данная группа теперь должна получать особое внимание при наблюдении в стационере и вне его. Стоит отметить, что критерии диагностики АК 1994 года, исключали это [46].
На современном этапе развития технологий, всё же, нельзя исключить в полной мере генетическую предрасположенность пациентов с любой формой АК к данному заболеванию, но несмотря на это, однозначно конкретная этиологическая и патофизиологическая принадлежность АК до сих пор не выявлена. Стоит особое внимание уделять пациентам с аритмиями не ясного генеза, а также обследованию людей, занимающихся спортом, акцентируя на профессиональных атлетов, для профилактики внезапной сердечной смерти и снижения рисков различных проявлений сердечных патологий.