

Одной из самых опасных и сложных в плане терапевтического течения групп заболеваний являются наследственные заболевания. Спинально мышечная атрофия (СМА)- одно из таких, это редкое генетическое заболевание, для которого характерна постепенная дегенерация α-моторных нейронов передних рогов спинного мозга.
Пациенты СМА гомозиготны по дефектному гену (SMN1 в 95-98% случаев), то есть для его проявления необходимо, чтобы оба родителя являлись носителями рецессивного генетического нарушения. Несмотря на то, что частота встречаемости данной патологии — один случай на 6–10 тыс. человек, считается, что каждый 40–50-й житель Земли является носителем дефектного гена SMN1, а вероятность наличия данного заболевания у детей в семье двух носителей мутировавшего гена составляет 25%, что является несомненным показателем актуальности проблематики СМА. Ситуацию также усугубляет тот факт, что существующее лечение не имеет необходимой терапевтической силы и скорее носит паллиативный характер, имея при этом чрезвычайно высокую цену.
На данный момент самым дорогим препаратом не только из СМА-категории, но и из всех остальных, является новый лекарственный препарат «Золгенсма» (Zolgensma, онасемноген абепарвовек), цена которого составляет 2,125 млн долларов.
Как правило, СМА связана с потерями участка пятой хромосомы или точечными мутациями гена SMN1 (survival motor neuron, или ген выживаемости мотонейронов). Этот ген отвечает за синтез белка SMN, который определяет корректное считывание информации с участка мРНК, кодирующего в себе место доставки этой самой мРНК для начала синтеза с неё белка. При нехватке белка SMN механизм нарушается, а поскольку спинальные моторные нейроны обладают очень длинными аксонами, то нарушение транспорта мРНК становится для них критичным. Из-за того, что мРНК не достигает концов аксонов, наступает гибель нейронов, и, как следствие, последующая мышечная атрофия.
Онасемноген абепарвовек (onasemnogene abeparvovec, AVXS-101) представляет собой генотерапевтическое лечение, после единственной внутривенной дозы обеспечивающее замену отсутствующего или дефектного гена SMN1 на его функциональную копию. Итогом становится нормальная выработка белка выживаемости мотонейронов (SMN) — и соответствующее исцеление спинальной мышечной атрофии.
Генотерапия при помощи онасемногена абепарвовека предполагает аденовирусную доставку в организм трансгена SMN (представлен самокомплементарной двухцепочечной молекулой), кодирующего полностью функциональный белок SMN и встраивающегося в ядра моторных нейронов. Благодаря способности капсида нереплицирующегося рекомбинантного аденоассоциированного вирусного вектора серотипа 9 (AAV9) пересекать гематоэнцефалический барьер подтверждена экспрессия SMN в мотонейронах во всех отделах головного и спинного мозга. Использование энхансера цитомегаловируса и куриного бета-актина в качестве гибридного промотора определяет быструю и устойчивую экспрессию SMN.
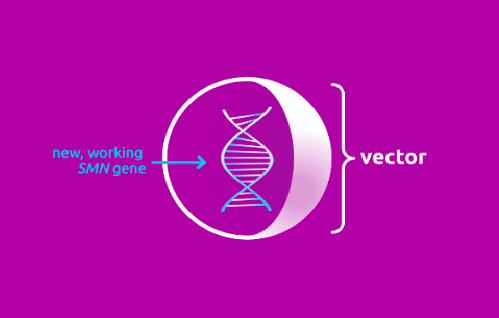
После того, как ген прибывает в нужную локацию, вектор разрушается и выводится из организма.
Оценка биораспределения трансгенных ДНК и мРНК и белка SMN, осуществленная цифровой капельной полимеразной цепной реакцией (ddPCRTM), полимеразной цепной реакцией с обратной транскрипцией (RT-PCR) и иммуногистохимическим окрашиванием соответственно, выявила, что векторные геномы, РНК-транскрипты и SMN-белки обнаруживаются во всех изученных отделах спинного мозга, в том числе шейном, грудном, поясничном и крестцовом. Экспрессия SMN-белка в моторных нейронах спинного мозга установлена на уровне, схожем с таковым для тканей, не пораженных спинальной мышечной атрофией типа I.
Анализ холинацетилтрансферазы (ChAT), маркера моторных нейронов, засвидетельствовал изобилие последних, причем с нормальными размерами и формой.
В общем и целом доказано, что однократное внутривенное введение «Золгенсма» способно восстановить SMN-экспрессию в моторных нейронах, лишенных функционального гена SMN1. Онасемноген абепарвовек разработан с тем прицелом, что спинальная мышечная атрофия является моногенным заболеванием, то есть достаточно доставить в организм корректную копию проблемного гена, чтобы остановить прогрессирование патологии.
Однако, невзирая на достаточно высокую эффективность «Золгенсма», препарат имеет целый ряд существенных недостатков. В первую очередь это, конечно же, высокая стоимость (2,125 млн долларов). Возможными побочными эффектами препарата являются: рвота, повышение уровня аминотрансфераз, тромбоцитопения, нарушение функций печени (вплоть до острого тяжёлого поражения). Не рекомендуется использование «Золгенсма» у недоношенных детей до достижения ими полного гестационного возраста.
При клинических исследованиях ввиду редкости заболевания СМА была небольшая выборка пациентов, а это означает, что количество побочных эффектов препарата может быть значительно выше, чем известно на данный момент. Важно также отметить, что долгосрочное влияние препарата на организм человека пока неизвестно. Прежде всего, не ясно, будет ли экспрессия гена SMN1 в организме пациента поддерживаться постоянно или постепенно сойдет на нет. Также не проводились исследования на животных по оценке онкогенного и мутагенного действий препарата.
Таким образом, «Золгенсма» может оказаться как настоящей панацеей для людей, больных СМА, так и генетической бомбой замедленного действия, эффект которой будет заметен только через годы или десятилетия.
Как бы то ни было, сегодня «Золгенсма» является одним из немногих одобренных генотерапевтических препаратов, и единственным — для лечения СМА. Данная технология является чрезвычайно перспективной и теоретически может подарить шанс СМА-пациентам на продолжительную жизнь высокого качества. Однако возможные побочные эффекты и высокая стоимость препарата пока не позволяют делать поспешных радужных выводов.
Библиографическая ссылка
Шайгородский А.А., Митюшникова Е.Б. ИСПОЛЬЗОВАНИЕ "ЗОЛГЕНСМА" В ЛЕЧЕНИИ СПИНАЛЬНО МЫШЕЧНОЙ АТРОФИИ // Международный студенческий научный вестник. 2021. № 1. ;URL: https://eduherald.ru/ru/article/view?id=20408 (дата обращения: 06.04.2025).