

Введение
Ожирение - хроническое заболевание, которое стремительно распространяется среди населения. Оно затрагивает не только взрослых, но и детей, подростков. Это связано с тяжёлыми метаболическими нарушениями, растущей сердечно-сосудистой заболеваемостью [1]. Жировая ткань вырабатывает огромное количество гормонов и цитокинов, регулирующих метаболизм, и в то же время способных глубоко и негативно воздействовать на физиологию эндотелия [4]. Это состояние может привести к формированию атеросклеротической бляшки [5].
Состояние хронического воспаления при ожирении вызывает нарушение регуляции эндокринных и паракринных эффектов продуктов адипоцитов, которые нарушают сосудистый гомеостаз и способствуют нарушению функции эндотелиальной вазодилатации и последующей гипертонии [2]. Здоровая периваскулярная жировая ткань (далее - ПВЖТ) обеспечивает вазодилатацию, в то время как ПВЖТ в состоянии ожирения приводит к изменению профиля высвобождаемых адипоцитокинов. Итогом служит снижение вазодилатирующего эффекта [24]. Воспаление жировой ткани, биодоступность оксида азота (далее - NO), инсулинорезистентность и окисленные липопротеины низкой плотности (далее - оЛПНП) являются основными факторами патогенеза в эндотелиальной дисфункции, сопутствующей ожирению [10][11]. Разрушение соединений между эндотелиоцитами, значительное увеличение продукции активных форм кислорода (далее - АФК), воспалительные медиаторы (которые берут своё начало из воспаленных эндотелиальных клеток), баланс между синтезом NO и АФК, передача сигналов инсулина и синтез NO, а также снижение соотношения L-аргинина/эндогенного асимметричного диметил-L-аргинина (далее - AДMA) также рассматриваются в связи с эндотелиальной дисфункцией при ожирении [11].
В данной работе раскрываются такие аспекты этой проблемы, как: физиология эндотелия; объясняются механизмы, благодаря которым ожирение (посредством продуктов секреции жировой ткани) влияет на функцию эндотелия; возможные патогенетические связи между ожирением и кардиоваскулярными заболеваниями, опосредованными через окислительный стресс, воспаление и эндотелиальную дисфункцию. Отдельно упоминается функция периваскулярной жировой ткани и её особенности у пациентов с ожирением.
Не менее пристально рассматривается роль гипероксии жировой ткани, гипоксического ответа и механизмы воздействия HIF-1a (фактор, индуцируемый гипоксией 1-альфа) на эти процессы, а также участие окисленных липидов низкой плотности в патогенезе эндотелиальной дисфункции.
Основная часть
ФИЗИОЛОГИЧЕСКАЯ РОЛЬ ЭНДОТЕЛИЯ
Эндотелий выстилает внутреннюю поверхность сосудов (кровеносных и лимфатических) и представлен плоскими клетками - эндотелиоцитами мезенхимного происхождения. Эндотелиоциты играют значительную роль в регуляции кардиоваскулярного гомеостаза и в поддержании здоровья сердечнососудистой системы [3]. Кроме формирования физического барьера между сосудистой стенкой и просветом сосуда, эндотелий секретирует медиаторы, регулирующие агрегацию тромбоцитов, коагуляцию, фибринолиз и тонус сосуда. Эти медиаторы также могут оказывать вазоконстрикторное действие (эндотелин-1, тромбаксан А2) и вазодилатирующее действие (оксид азота, простациклин, эндотелиальный гиперполяризационный фактор) [2].
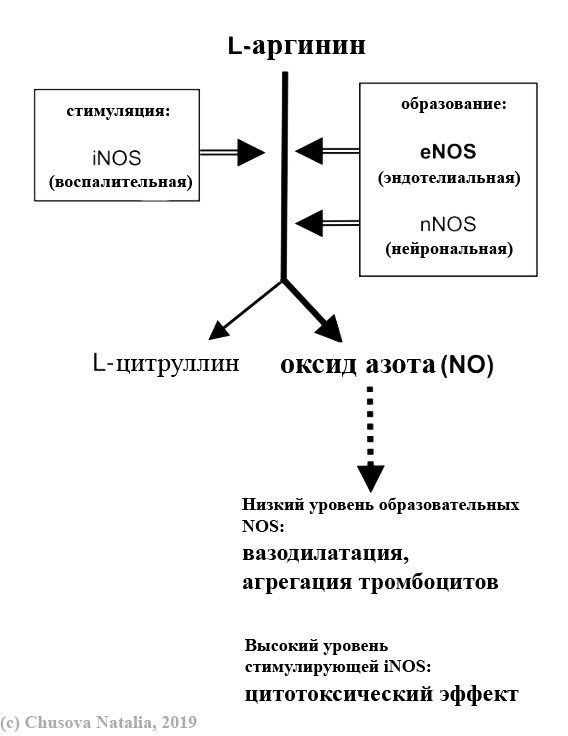
Эндотелий-зависимая вазодилатация крупных сосудов во многом ассоциирована с действием оксида азота, в то время как в сосудах меньшего диаметра преобладает эндотелиальный гиперполяризационный фактор [41]. Предшественник оксида азота – L-аргинин. Внутриклеточный L-аргинин может превращаться в L-цитруллин и оксид азота, либо в L-орнитин и мочевину. Превращению в оксид азота способствует группа ферментов: NO-синтазы. Существуют три изоформы: эндотелиальная (eNOS), нейрональная (nNOS) и воспалительная (iNOS) NO-синтазы [17]. Эти ферменты катализируют НАДФН и O2-зависимое окисление L-аргинина до NO и цитруллина [см. Приложение 1]. NOS включают в себя гемсодержащий и флавиновый участки, и активны только в качестве димеров. Димеризация активирует фермент путем секвестрации железа, генерируя высокоаффинные участки связывания для аргинина и необходимого кофактора тетрагидробиоптерина (BH4) и позволяя переносить электроны из флавинов редуктазного домена в гемоксигеназный домен. Активность также зависит от связанного кальмодулина. Кроме того, активность eNOS также может регулироваться посттрансляционными модификациями: эти модификации происходят посредством фосфорилирования Ser1179, что повышает активность фермента. Это звено могут фосфорилировать несколько киназ, включая протеинкиназу А, протеинкиназу С и серин/треонин-киназу Akt [14]. Миристоилирование и пальмитоилирование поддерживают локализацию eNOS в кавеолах (колбообразные впячивания плазматической мембраны в эндотелиоцитах), где eNOS связана с кавеолином, который сохраняет фермент неактивным [см. Приложение 2]. Активация эндотелиальных рецепторов ацетилхолина активирует фосфолипазу C (ФЛC), которая катализирует выработку инозитол-1,4,5-трифосфата (IP3) и диацилглицерина (ДАГ) из фосфатидилинозитол-4,5-бифосфата (PIP2). IP3-индуцированное повышение внутриклеточного кальция активирует кальмодулин, связывающийся с eNOS, который диссоциирует от кавеолина и транслоцируется в цитоплазму. Фосфорилирование eNOS протеинкиназой A (ПКА) инактивирует фермент, который затем перемещается в мембрану кавеолина.
Показано, что инсулин обладает вазодилирующим действием: этот эффект обеспечивается тем, что этот гормон может стимулировать синтез NO in vivo, посредством активации пострецепторных путей, которые включают фосфатидилинозитол-3-киназу (PI3K) и киназы Akt [46]. В резистентных к инсулину состояниях, таких как ожирение, изменения пострецепторных путей инсулина нарушают не только метаболические, но и сосудистые эффекты гормона. NO-чувствительная гуанилилциклаза (NO-чувствительная ГК) является наиболее важным рецептором для сигнальной молекулы NO. Последний стимулирует выработку циклического гуанозинмонофосфата (цГМФ) путем активации растворимой ГК. цГМФ регулирует большинство своих внутриклеточных эффектов посредством активации специфических цГМФ-зависимых протеинкиназ (ПКG). Несколько семейств фосфодиэстераз (ФДЭ-I-VI) действуют как регуляторные переключатели, катализируя деградацию цГМФ до гуанозин-5V-монофосфата (5V-ГМФ). Сигнальный каскад NO/цГМФ имеет решающее значение в сердечно-сосудистой системе, где он контролирует расслабление гладких мышц и подавление агрегации тромбоцитов; циклические нуклеотидные ФДЭ гидролизуют цГМФ и тем самым прекращают свое действие. Следует отметить недавние сообщения, что у мышей с дефицитом лептина, страдающих ожирением, сигнальный путь NO/цГМФ значительно изменен в желудочковых миоцитах [37].
NO оказывает несколько важных воздействий на сосудистую сеть. Во-первых, он поддерживает тонус, расслабляя клетки гладких мышц сосудов; он также ингибирует адгезию, активацию, секрецию и агрегацию тромбоцитов и способствует дезагрегации тромбоцитов. В дополнение к этим эффектам NO, происходящий из эндотелия, подавляет адгезию лейкоцитов к эндотелию, миграцию и пролиферацию гладкомышечных клеток: следовательно, NO является мощным ингибитором всех этих механизмов, которые в конечном итоге приводят к пролиферации и атеросклерозу. Эндотелий способствует регуляции артериального давления и кровотока, высвобождая не только NO, но и несколько других соединений, которые способствуют как вазодилатации, так и вазоконстрикции [6].
Кроме того, эндотелий производит соединение, известное как EDHF (эндотелиальный гиперполяризационный фактор, далее - EDHF), которое способствует расслаблению и расширению сосудов гладкой мускулатуры, активируя АТФ-чувствительные калиевые каналы и натриево-калиевую АТФазу гладких мышц. Точная природа EDHF, однако, остается неясной [8].
ЖИРОВАЯ ТКАНЬ, АДИПОКИНЫ И ЭНДОТЕЛИЙ
Жировая ткань может производить значительное количество соединений, способных влиять на функцию эндотелия, наиболее важными из которых являются лептин, резистин, фактор некроза опухоли альфа (ФНОа), интерлейкин-6 (IL-6), белок-хемоаттрактант моноцитов-1 (MCP-1), ингибитор активатора плазминогена-1 (ИАП-1), адипонектин и белки ренин-ангиотензиновой системы [33]. Последние данные показывают, что существует взаимодействие между секреторными белками адипоцитов, называемыми адипокинами, и эндотелием; таким образом, способность адипокинов непосредственно влиять на сосудистый гомеостаз может представлять собой одну из причин сердечно-сосудистых заболеваний у пациентов с ожирением [30].
Лептин представляет собой белок из 167 аминокислот, который экспрессируется главным образом адипоцитами и высвобождается в крови пропорционально размерам жировой ткани; действие лептина в ЦНС способствует снижению веса за счет снижения потребления пищи и увеличения расхода энергии. Недавние исследования показали, что лептин обладает широким спектром влияния на гомеостаз сосудов, он в том числе влияет на функцию эндотелия. Исследования in vitro показали, что лептин вызывает окислительный стресс в культивируемых эндотелиальных клетках, увеличивая образование активных форм кислорода (АФК). Также было показано, что лептин стимулирует секрецию провоспалительных цитокинов (например, фактор некроза опухоли А, интерлейкин-6), которые, как известно, не только способствуют гипертонии, но также влияют на функцию эндотелия. Однако, лептин может иметь прямые сосудистые эффекты, которые приводят к снижению артериального давления. Вазорелаксация, вызванная лептином, является гетерогенной и связана с преобладающей ролью механизмов EDHF. Однако другие исследования продемонстрировали, что лептин может вызывать вазодилатацию посредством стимуляции NO. Существует предположение, что вызванное лептином высвобождение NO определяется не только прямым воздействием на эндотелий сосудов, но и косвенным воздействием на адипоциты: эти клетки под контролем лептина действительно могут играть главную роль в высвобождении NO путем активации NO-синтазы [11].
Резистин - это адипокин, который, как полагают, играет роль в развитии инсулинорезистентности и ожирения. Резистин, по-видимому, продуцируется во время адипогенеза и ингибирует поглощение глюкозы клетками скелетных мышц на животных моделях эксперимента. Инкубация эндотелиальных клеток с человеческим рекомбинантным резистином приводит к увеличению высвобождения эндотелина-1 без изменения продукции NO. В дополнение к этому, обнаружено, что обработанные резистином клетки показали повышенную экспрессию сосудисто-клеточных молекул адгезии (VCAM) -1 и MCP-1 [42]. Эти данные свидетельствуют о том, что резистин непосредственно активирует эндотелиальные клетки, способствуя высвобождению эндотелина-1 и активируя молекулы адгезии. Тем не менее, необходимы дальнейшие исследования, чтобы определить биологическую значимость резистентных сосудистых эффектов in vivo у людей.
Адипонектин - полипептид с молекулярной массой 30 кДа, высоко и специфически экспрессируемый в дифференцированных адипоцитах, циркулирует в кровотоке. Установлена ??прочная обратная связь между адипонектином, инсулинорезистентностью и воспалительными состояниями. Внутри сосудистой стенки адипонектин оказывает несколько эффектов, которые опосредуются повышенным фосфорилированием рецептора инсулина, активацией АМФ-активированной протеинкиназы (АМФК) и модуляцией пути транскрипционного фактора NF-κB. Исследования in vitro показали, что он ингибирует адгезию моноцитов за счет снижения экспрессии молекул адгезии, ингибирует превращение макрофагов в пенистые клетки и уменьшает пролиферацию мигрирующих гладкомышечных клеток в ответ на факторы роста. Адипонектин также обладает способностью стимулировать выработку NO в эндотелиальных клетках, используя фосфатидилинозитол-3-киназозависимые пути, включающие фосфорилирование eNOS в Ser1179 с помощью АМФК. Кроме того, адипонектин также может вызывать ангиогенез, стимулируя перекрестные связи между АМФ-активированной протеинкиназой и передачей сигналов Akt в эндотелиальных клетках. Таким образом, адипонектин обладает сильными антиатерогенными свойствами, которые были подтверждены также in vivo у людей. Ouchi и соавт. проанализировали функцию эндотелия у 202 пациентов с артериальной гипертензией и обнаружили, что уровень адипонектина в плазме сильно коррелирует с ответом на расширение сосудов на реактивную гиперемию, явление, опосредованное NO. Выявлена прямая связь между уровнями адипонектина в плазме и эндотелий-зависимой вазодилатацией на микроциркуляторном уровне в предплечье у пациентов с гипертонической болезнью [34]. Поскольку ожирение характеризуется снижением выработки и активности адипонектина [21], возможно, что в таких клинических условиях возникает дисбаланс по отношению к тем факторам, которые способствуют развитию эндотелиальной дисфункции и атеросклеротического процесса [32].
МСР-1 (моноцитарный хемотаксический фактор-1) представляет собой хемокин, который привлекает моноциты в места воспаления, экспрессируется и секретируется жировой тканью. Жировая ткань может быть основным источником повышенных уровней MCP (в 10-100 раз больше, чем в печени) в плазме, наблюдаемых у мышей с ожирением. МСР-1 оказывает прямое ангиогенное действие на эндотелиальные клетки: было замечено, что МСР-1 ускоряет заживление ран, процесс, который зависит от роста кровеносных сосудов [38]. Несмотря на растущее количество информации, влияние MCP-1 на функцию эндотелия при ожирении у человека полностью не раскрыто.
РОЛЬ ВОСПАЛЕНИЯ
Все больше фактов свидетельствует о том, что жировая ткань в целом и висцеральное ожирение в частности являются ключевыми регуляторами воспаления. Жировая ткань секретирует провоспалительные цитокины, такие как ФНОa и IL-6, которые, по-видимому, играют важную роль в воздействии как на функцию эндотелия, так и на метаболизм глюкозы [33]. Все больше доказательств указывает на причинную связь между воспалением и инсулинорезистентностью. ФНОa опосредует резистентность к инсулину в результате ожирения во многих моделях ожирения у грызунов [28]. Кроме того, было показано, что МСР-1 также снижает чувствительность адипоцитов к инсулину [38]. Что наиболее важно, инфильтрация макрофагами жировой ткани при ожирении может быть неотъемлемой частью этих воспалительных изменений. Некоторые воспалительные реакции имели место вне адипоцитов, в макрофагах, инфильтрирующих расширяющуюся жировую ткань. Тем не менее, важнейшие вопросы о механизмах, с помощью которых воспалительный ответ запускается и поддерживается при ожирении: являются ли адипоциты самими антигенами? Или воспалительный ответ происходит не по классической схеме: реакции антиген-антитело? Или это физическое повреждение эндотелия, вызванное несколькими факторами риска сердечно-сосудистых заболеваний?
Каким бы ни был первичный стимул, провоспалительные цитокины негативно влияют на эндотелий. ФНОa, трансмембранный белок с молекулярной массой 26 кДа, который оказывает свое действие через рецепторы ФНОa типа I и типа II, не только индуцирует инсулинуорезистентность, но и глубоко влияет на функцию эндотелия. ФНОa стимулирует активацию ядерного фактора транскрипции каппа B (NF-kB); NF-kB играет критическую роль во влиянии на воспалительные реакции и апоптоз: он также регулирует экспрессию факторов роста, провоспалительных цитокинов и молекул адгезии. Многие продукты генов, регулируемых NF-kB, также, в свою очередь, активируют NF-kB (в том числе ФНОa). Благодаря этой активации ФНОa индуцирует окислительный стресс, который усугубляет патологические процессы, приводящие к эндотелиальной дисфункции и атерогенезу [2]. Было показано, что усиленное производство ФНОa увеличивает активность индуцируемого NOS, то есть фермента, который продуцирует NO в большом количестве, является кардиотоксичным и способствует апоптозу. ФНОa опосредует повышенную проницаемость эндотелия путем активации НАДФН-оксидазы. Наконец, ФНОa ингибирует транскрипционную, а также посттранскрипционную экспрессию гена eNOS, что позволяет объяснить эндотелиальную дисфункцию. ФНОa ингибирует индуцированное инсулином увеличение экспрессии e-NOS в эндотелиоцитах аорты человека. Посредством этого механизма ФНОa может воздействовать на неспособность инсулина вызывать вазодилатацию при ожирении и сахарном диабете 2 типа. Помимо этого, отчеты экспериментов указывают на то, что ФНО-α может стимулировать выработку АФК посредством активации НАДФН-оксидазы или активации ядерного транскрипционного фактора-каппа B (NF-kB), который, в свою очередь, опосредует экспрессию воспалительных цитокинов [9][45].
IL-6 является другим цитокином, связанным с ожирением и инсулинорезистентностью. Он циркулирует в кровотоке в нескольких гликозилированных формах размером от 22 до 27 кДа. IL-6 и его рецептор экспрессируются адипоцитами и матрицей жировой ткани: до трети циркулирующего IL-6 происходит из жировой ткани. IL-6 отрицательно влияет на функцию эндотелия; он является важным медиатором повышенной эндотелиальной проницаемости, влияя на неё через изменения ультраструктурного распределения плотных соединений и морфологические изменения в форме клеток [45]. Протеинкиназа C (ПКC) является важным внутриклеточным мессенджером в этих IL-6-опосредованных изменениях. Также было показано, что IL-6 может индуцировать эндотелиальную дисфункцию, активируя рецептор ангиотензина II AT1: этот эффект может также способствовать отражению окислительного стресса, вызванного провоспалительным цитокином при ожирении [40].
Повышенная концентрация С-реактивного белка в плазме (СРБ), маркер низкого уровня хронического воспаления, связана с метаболическим синдромом. Поскольку жировые отложения в брюшной полости являются источником IL-6, который в значительной степени стимулирует синтез СРБ печенью, абдоминальное ожирение является важным фактором, помогающим объяснить воспалительную реакцию при ожирении. Доказана связь между уровнями СРБ в плазме и всеми показателями ожирения, такими как ИМТ, общая масса жира тела и обхват талии. Повышенная концентрация СРБ важна, поскольку он, помимо того, что является маркером воспаления, может также непосредственно способствовать дисфункции эндотелия. Воздействие эндотелиальных клеток на СРБ снижает выработку эндотелиального NO и снижает экспрессию eNOS из-за снижения стабильности мРНК eNOS [45].
Таким образом, повышенная выработка цитокинов, возникающая из-за увеличения абдоминального жира, может быть ответственна не только за метаболические нарушения, связанные с синдромом инсулинорезистентности, но также за повышенный риск сердечно-сосудистых заболеваний, наблюдаемый у пациентов с абдоминальным ожирением.
РОЛЬ ПЕРИВАСКУЛЯРНОЙ ЖИРОВОЙ ТКАНИ (ПВЖТ)
Всё больше данных указывают на то, что ПВЖТ играет непосредственную роль в регуляции воспаления слабой степени, также секретируя ФНО-a и IL-6. Было доказано, что здоровая ПВЖТ секретирует факторы, которые влияют на расширение сосудов путем увеличения биодоступности NO, этот эффект был утрачен в ПВЖТ у пациентов с ожирением. Введение ФНО-a и IL-6 в ПВЖТ вокруг здоровых кровеносных сосудов снижало активность процесса расширения. Эти изменения были отменены веществами, поглощающими АФК или инкубацией антагонистов цитокинов [23]. Данные результаты свидетельствуют о том, что вазопротективный эффект здоровой ПВЖТ теряется при ожирении из-за развития гипертрофии адипоцитов, приводиящей к гипоксии, окислительному стрессу и усилению накопления ФНО-a. Эндотелиальная дисфункция, связанная с ожирением, включает сосудистый дисбаланс ET (эндотелин)-1/NO в пользу патологической активации эндогенной системы ET-1 [43]. Предполают, что избыток АФК (посредством активации НАДФН-оксидазы) индуцирует расцепление связанной eNOS, что, в свою очередь, способствует образованию супероксида и снижению выделения NO. В недавнем исследовании обнаружено, что сосуды людей, страдающих ожирением, показали малоактивное эндотелий-зависимое расслабление (вызванное снижением доступности NO). Результаты исследования мелких сосудов у пациентов с ожирением показывают вторичность сниженной биодоступности NO по отношению к избыточной выработке АФК. ФНО-а способствует эндотелиальной дисфункции, стимулируя образование внутрисосудистых АФК. Кроме того, иммуногистохимия показала заметную активацию ФНО-a в медиальном слое этих сосудов, подтверждая гипотезу о том, что сосудистая стенка является источником ФНО-a, участвующего в дисфункции эндотелия [44].
ГИПЕРОКСИЯ ЖИРОВОЙ ТКАНИ
При ожирении избыточность жировой ткани связана с гипетрофией и гиперплазией адипоцитов, в то время как наполнение капилляров не отвечает требованиям роста жировой ткани. Барьерный эффект крупных адипоцитов, скорее всего, способствует изменениям кровотока и парциального давления кислорода в жировой ткани [47]. Парциальное давление кислорода (рО2) в межклеточном пространстве между адипоцитами людей с ожирением выше, чем у худых, несмотря на уменьшенный кровоток в жировой ткани. Людям с ожирением свойственна гипероксия жировой ткани, несмотря на сниженный кровоток в ней. Этот парадокс объясняется снижением потребления кислорода в жировой ткани в сочетании с инсулинорезистентностью, нарушением васкуляризации жировой ткани и повышенным воспалением жировой ткани. Тем не менее, результатом этих событий является местный гипоксический ответ [22]. Гипоксический ответ в адипоцитах и макрофагах является одной из важных причин хронического воспаления при висцеральном ожирении. Следовательно, повышенная активность HIF-1a (фактор, индуцируемый гипоксией 1-альфа) является признаком хронического воспаления в жировой ткани при развитии ожирения [25].
Адипогенез приводит к увеличению экспрессии фактора роста эндотелия сосудов (далее – VEGF – от англ.VascuLar endothelial growth factor) через HIF-1альфа. В то же время гипоксия адипоцитов может привести к экспрессии гена путем активации NF-kB. Собственно пролиферация эндотелиальной клетки в первую очередь зависит от проангиогенного фактора VEGF (фактор роста эндотелия сосудов). Кроме того, активность тромбоцитарного фактора роста (platelet-derived growth factor, PDGF) в ангиогенезе зависит от активности VEGF. PDGF в сыворотке стимулирует дифференциацию эндотелиальных клеток и выделяется многими типами клеток, включая тромбоциты, макрофаги, фибробласты и эндотелиальные клетки. PDGF экспрессируется во всех типах клеток, составляющих жировую ткань, однако уровни экспрессии различаются. Преадипоциты экспрессируют больше PDGF, чем зрелые адипоциты. При ожирении количество преадипоцитов уменьшается, так как большинство из них дифференцируются в зрелые адипоциты. Чтобы восполнить пул PDGF, инфильтрация макрофагов в жировую ткань увеличивается для компенсации потерь преадипоцитов [35]. Учитывая все эти данные, при ожирении макрофаги могут служить стимуляторами ангиогенеза в жировой ткани. Тем не менее, PDGF, VEGF и эндотелиальные клетки необходимы для формирования капилляров. Таким образом, количество HIF-1 увеличивается.
РОЛЬ ОКСИЛЕННЫХ ЛИПОПРОТЕИНОВ НИЗКОЙ ПЛОТНОСТИ
В исследованиях была показана прочная связь оЛПНП со всеми компонентами метаболического синдрома и С-реактивным белком. (Holvoet et aL. 2008b). Лечение эндотелиальных клеток с оЛПНП стимулирует связывание моноцитов, а также производство хемотаксических факторов для моноцитов. Индукция экспрессии гранулоцитарно-макрофагального колониестимулирующего фактора (GM-CSF), M-КСФ и G-КСФ влияют на миграцию и пролиферацию эндотелиоцитов. Кроме того, оЛПНП способствует гиперплазии и гипертрофии адипоцитов, обеспечивая высокую скорость пролиферации этих клеток, низкий уровень апоптоза и нарушение процесса дифференцировки с увеличением экспрессии мРНК пре-адипоцитарного фактора-1.
Белок острой фазы (СРБ) специфически связывается с оЛПНП посредством связывания с Fc-гамма-рецепторами на макрофагах, что может привести к увеличению поглощения оЛПНП макрофагами. При повышенных уровнях ЛПНП в плазме этот механизм может способствовать воспалению эндотелия, образованию пенистых клеток и ускорению развития атеросклероза.
В физиологических условиях существует корреляция между снижением экспрессии мРНК NOS и угнетением активности NO-синтазы, висцеральным ожирением, транскрипцией мРНК eNOS. Ее деградация независимо регулируется оЛПНП. В то время как неизменённые ЛПНП могут ингибировать выработку NO путем снижения экспрессии белка NOS или ослабления его ферментативной активности, воздействие нецитотоксических концентраций оЛПНП вызывает прогрессирующее снижение уровней мРНК NOS. Подавление экспрессии eNOS в ответ на атерогенные концентрации неизменённых ЛПНП потенциально является механизмом нарушения функции эндотелия. ОЛПНП-индуцированное перераспределение eNOS из кавеол путем смещения eNOS и кавеолина-1 во внутриклеточный компартмент вызывает последующую неспособность активировать eNOS с помощью ацетилхолина. Что касается упомянутых выше механизмов, повышение оЛПНП стремительно ослабляет способность к синтезу NO эндотелием и вызывает эндотелиальную дисфункцию. Изменённые частицы ЛПНП также индуцируют эндотелиальную секрецию хемотаксических веществ и экспрессию рецепторов адгезии, которые способствуют активации, адгезии и перемещению моноцитов и Т-клеток в артериальную стенку [11].
Моноцитарный комплекс MCP-1/CCL2 взаимодействует с моноцитарным рецептором CCR2. Последующее накопление моноцитов в эндотелиальном слое способствует их проникновению путем диапедеза. Перемещение моноцитов происходит в областях, где базальная пластинка обогащена изменёнными частицами ЛПНП. Это происходит, главным образом, через его зону контакта между эндотелиальными клетками. Соединительные молекулы адгезии -A и -C участвуют в контроле проницаемости сосудов и перемещении лейкоцитов через мембраны эндотелиальных клеток. Таким образом, ЛПНП модифицируют антитромботические свойства эндотелия сосудов и влияют на сократимость сосудов, уменьшая биодоступность эндотелиального NO и активируя провоспалительные сигнальные пути [12]. Как известно, субэндотелиальное накопление пенистых клеток, происходящих из макрофагов, является одной из характерных черт атеросклероза. Таким образом, инкубация совместных культур эндотелиальных клеток аорты человека и клеток гладких мышц с ЛПНП приводила к 7,2-кратной индукции мРНК для MCP-1 и 7,1-кратному увеличению переноса моноцитов в субэндотелиальное пространство культур [11].
Накопление моноцитов в интиме требует взаимодействия локально продуцируемых хемокинов со специфическими поверхностными рецепторами клетки, включая рецептор CCR2 на MCP-1. Повышению экспрессии CCR2 и хемотаксиса способствуют неизменённые ЛПНП, но не оЛПНП. Фактически, оЛПНП быстро подавляют экспрессию CCR2. Напротив, повышенные уровни ЛПНП в плазме усиливают экспрессию CCR2, хемотаксический ответ и потенциально способствуют увеличению привлечения моноцитов в стенку сосуда при хроническом воспалении и атерогенезе. Как только моноциты достигают интимы, колониестимулирующие факторы побуждают их фенотипически превращаться в макрофаги. Они начинают поглощать изменённые частицы ЛПНП. Полностью окисленный ЛПНП вызывает адгезию моноцитов [11]. Этот механизм может дополнительно объяснить атерогенный потенциал оЛПНП.
ОЖИРЕНИЕ, ОКИСЛИТЕЛЬНЫЙ СТРЕСС И ЭНДОТЕЛИАЛЬНАЯ ДИСФУНКЦИЯ
Окислительные реакции имеют решающее значение во всех событиях, которые приводят к атерогенезу, включая эндотелиальную дисфункцию. Влияние свободных радикалов (АФК), происходящих из кислорода, на сосудистую функцию в большой степени зависит от их количества. При образовании в небольших количествах они могут действовать как внутриклеточные вторичные мессенджеры, стимулируя ответы в виде роста гладкомышечных клеток сосудов и фибробластов. Более высокие количества АФК могут вызвать широко распространенную цитотоксичность [36]. Практически все типы клеток сосудов продуцируют АФК, которые могут регулировать такие субстанции, как молекулы адгезии и хемотаксические факторы, антиоксидантные ферменты и вазоактивные вещества. Повышенная регуляция молекул адгезии и хемотаксических молекул с помощью чувствительных к окислению механизмов имеет особое значение для эндотелиальной дисфункции, поскольку эти молекулы способствуют адгезии и миграции моноцитов в стенку сосуда. В эндотелии, подверженном воздействию агентов, которые повреждают сосудистую сеть, происходит стимуляция нескольких ферментов, которые могут продуцировать АФК: ферменты митохондриальной цепи переноса электронов, ксантиноксидаза, циклооксигеназы, липооксигеназы, миелопероксидазы, цитохром P450 монооксигеназы, несвязанные NOS, гемоксигеназы, пероксидазы и НАДФН-оксидазы. АФК могут продуцироваться внутриклеточно, внеклеточно или в определенных внутриклеточных отделах. Среди этих ферментов никотинамид-аденин-динуклеотид/НАДФН-оксидаза является актуальным, поскольку является основным сосудистым источником АФК. Между НАДФН-оксидазной активностью, атеросклеротическими факторами риска и эндотелиальной дисфункцией существует прочная корреляция. Циркулирующие лимфомоноциты от пациентов с диабетом 2 типа являются участками окислительного стресса, экспрессия гена НАДФН-оксидазы повышена и это увеличение зависит от метаболического контроля [7]. Примечательно, что активность НАДФН увеличивается не только за счет факторов, которые повреждают сосудистый эндотелий, но, возможно, за счет самого инсулина и макроэлементов. Например, тест на толерантность к глюкозе приводит к значительному увеличению выработки АФК у нормальных субъектов со специфическим приростом экспрессии p47phox, ключевого белкового компонента НАДФН-оксидазы [39]. Более того, накопление жира коррелирует с системным окислительным стрессом у людей и мышей. Продукция АФК селективно возрастала в жировой ткани мышей с ожирением и была связана с повышенной экспрессией НАДФН-оксидазы и снижением экспрессии антиоксидантных ферментов. Также, в культивируемых адипоцитах было показано, что повышенные уровни жирных кислот увеличивали окислительный стресс через НАДФН-оксидазу. Этот повышенный окислительный стресс вызывает нарушение регуляции выработки адипоцитокинов (гормонов жирового происхождения), включая адипонектин, ИАП-1, IL-6 и MCP-1 [15]. В исследованиях было показано: у людей с ожирением увеличиваются повреждения, вызванные активными формами кислорода, в липидах, белках и аминокислотах [10]. В частности, перекисное окисление липидов, благодаря производству биоактивных изо-эйкозаноидов, может усиливать и поддерживать не только слабое системное воспаление, но и активацию тромбоцитов у женщин с ожирением по мужскому типу. Повышенный окислительный стресс усиливает разрушение оксида азота, тем самым снижая его биологический эффект.
Другие факторы, связанные с ожирением и резистентностью к инсулину, такие как свободные жирные кислоты и низкие концентрации липопротеинов высокой плотности (ЛПВП), также усиливают окислительный стресс, способствуя снижению биодоступности оксида азота [19]. Повышенный окислительный стресс, по-видимому, является основным механизмом, посредством которого инсулинорезистентность вызывает эндотелиальную дисфункцию. Предполагается, что резистентность к инсулину как таковая, независимо от гипергликемии, может способствовать выработке АФК. Другим источником свободных радикалов является так называемая несвязанная eNOS, состояние, при котором этот фермент лишен L-аргинина или тетрагидробиоптерина (BH4), важного кофактора для нормальной активности eNOS, что приводит к образованию O2 и H2O2 вместо NO. Разобщение происходит при эндотелиальной дисфункции, что приводит к снижению биодоступности NO, увеличению продукции O2 и образованию пероксинитрита (ONOO-), ключевого медиатора перекисного окисления липидов и образования пенистых клеток при атеросклеротических поражениях [см. Приложение 2]. Можно предположить, что нефизиологическое повышение активности этого фермента может привести к усилению окислительного стресса из-за несвязанного eNOS [18].
АФК могут генерироваться внутриклеточно и внеклеточно несколькими ферментами. Разобщение eNOS способствует производству АФК. Цитокины, выделяемые жировой тканью, могут усиливать окислительное повреждение.
Важно, что уровни супероксида в сосудах и активность NO определяются скоростью разложения супероксида. Основной ферментной системой, разрушающей свободные радикалы кислорода, является супероксиддисмутаза. Внеклеточная форма супероксиддисмутазы расположена между эндотелием и клетками гладких мышц сосудов. Экспериментально и клинически было показано, что ожирение связано со снижением антиоксидантного механизма: это состояние делает пациентов более склонными к окислительному стрессу [20].
Наконец, продукция NO может быть блокирована эндогенными ингибиторами. Асимметричный диметиларгинин (ADMA) является эндогенным конкурентным ингибитором связывания L-аргинина с eNOS и, следовательно, может принимать участие в нарушении регуляции реакции L-аргинин/NO. Повышенная продукция ФНО-a ингибирует распад ADMA: это может представлять важный механизм, посредством которого ожирение может изменить воздействия NO. Это было показано на пожилых мужчинах с высоким риском ССО, у которых была обнаружена тесная связь между ИМТ и уровнем ADMA в плазме крови, что, в свою очередь, указывает на связь с эндотелиальной дисфункцией у лиц с избыточным весом [16].
В совокупности эти данные свидетельствуют о том, что усиление окислительного стресса в накопленном жире играет основную роль в индукции эндотелиальной дисфункции при ожирении у человека [см. Приложение 3].
Заключение
У пациентов с ожирением множественные взаимосвязанные механизмы способствуют дисфункции эндотелиальных клеток. Данный контингент подвергается повышенному риску развития ишемической болезни сердца и её осложнений. Поэтому эндотелиальную дисфункцию необходимо либо предотвращать, либо корректировать путем изменения образа жизни или, если это не представляется возможным, направленной работой с каждым из факторов риска по-отдельности.
Продукты метаболизма жировой ткани различной локализации (лептин, резистин, адипокины и др.), играют важную роль в развитии воспаления слабой степени и эндотелиальной дисфункции. Нарушение передачи сигналов инсулина в мышечных, жировых и эндотелиальных клетках также усугубляет течение дисфункции эндотелия.
Циркуляция избыточного количества липидов является важной причиной как эндотелиальной дисфункции, так и инсулинорезистентности. Механизмы липид-опосредованной токсичности включают в себя: окислительный стресс, воспаление, дисфункцию митохондрий, стресс эндоплазматического ретикулума и, как итог, гибель клеток [36]. Тем не менее, очень важным аспектом эндотелиальной дисфункции (помимо цитотоксичности липидов) является снижение биодоступности NO, и, в дополнение, системные нарушения при ожирении, такие как гипергликемия, окислительный стресс, активация ренин-ангиотензиновой системы, повышение провоспалительных цитокинов, неадекватная вазодилатация или парадоксальная вазоконстрикция в коронарных и периферических артериях. Кроме того, дефицит NO является основным связующим звеном между инсулинорезистентностью и эндотелиальной дисфункцией.
С клинической точки зрения, терапевтические подходы, которые снижают: резистентность к инсулину, количество избыточных циркулирующих в крови липидов, окислительный стресс могут устранить эндотелиальную дисфункцию и снизить смертность от сердечно-сосудистых заболеваний. В связи с этим, изменения в образе жизни, способствующие похудению (в дополнение к фармакологическому лечению) являются полезными инструментами в борьбе с эндотелиальной дисфункцией, связанной с липотоксичностью. Небольшая потеря веса может улучшить эндотелиальную функцию и одновременно воздействовать на весь спектр факторов риска сердечно-сосудистых заболеваний.
Фармакологический подход в данном случае имеет решающее значение, поскольку определенные классы лекарств, такие как тиазолидиндионы, ингибиторы АПФ и статины, могут улучшать функцию эндотелия путем снижения окислительного стресса, повышения биодоступности оксида азота и уменьшения воспаления [13].
Приложение 1.
Рис. 1 Реакция образования NO из L-аргинина.
Приложениие 2.
Рис. 2 Физиология и патофизиология eNOS.
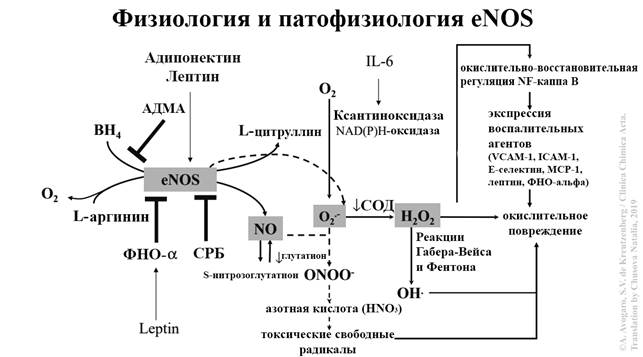
АДМА – асимметричный диметиларгинин. BH4 – тетрагидробиоптерин. О2 – кислород. eNOS - эндотелиальная NO-синтаза. НАДФН – оксидаза. IL-6 - интерлейкин-6. СРБ – С-реактивный белок. СОД – супероксиддисмутаза. Реакция Габера-Вейса - генерирует •OH (гидроксил-радикал) из H2O2 (пероксида водорода) и супероксида (•O2−). Реакция может вызывать окислительный стресс. Катализируется ионами железа. Реакция Фентона — реакция пероксида водорода с ионами железа. Продукты: Fe3+ + OH· + OH− . NF-каппа B - ядерный фактор-каппа B. Универсальный фактор транскрипции, контролирующий экспрессию генов иммунного ответа, апоптоза и клеточного цикла. VCAM-1 – васкулярная молекула клеточной адгезии. Белок, участвует в адгезии лейкоцитов и эндотелиальных клеток, передаче сигналов. ICAM-1 - молекула межклеточной адгезии-1. MCP-1 моноцитарный хемоатрактантный белок-1 - цитокин, относится к группе CC-хемокинов (β-хемокинов). Является наиболее мощным фактором хемотаксиса моноцитов.
Приложение 3.
Рис. 3 Механизм эндотелиальной дисфункции при ожирении.
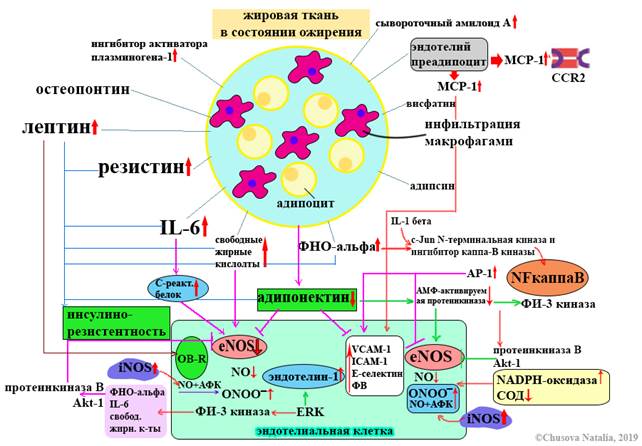
Выделяемые адипоцитами и воспалительными клетками высокоактивные молекулы проникают в жировую ткань. Помимо того, что все эти молекулы могут вызывать хроническое воспаление слабой степени, они также могут влиять на эндотелиальную функцию сосудов, регулируя баланс между синтезом NO и активными формами кислорода.
Ингибитор активатора плазминогена-1. сывороточный амилоид A. MCP-1 моноцитарный хемоатрактантный белок-1 - цитокин, относится к группе CC-хемокинов (β-хемокинов). Является наиболее мощным фактором хемотаксиса моноцитов. CCR2 (C-C рецептор хемокина 2) - рецептор β-хемокинов класса интегральных мембранных белков. CCR2 преимущественно отвечает за специфический хемотаксис моноцитов под влиянием MCP-1. IL-6 - интерлейкин-6. ФНО-альфа - фактор некроза опухоли-альфа. С-реактивный белок. Свободные жирные кислоты. (АМФ-)5 'аденозинмонофосфат-активируемая протеинкиназа. ФИ3-киназа - фосфоинозитид-3-киназа. Протеинкиназа В Акт 1- белок, кодируемый геном Акт1, участвует в регуляции клеточного цикла, апоптоза, метаболизма глюкозы и в процессе ангиогенеза. NO - оксид азота. eNOS - эндотелиальная NO-синтаза. VCAM-1 – васкулярная молекула клеточной адгезии. Белок, участвует в адгезии лейкоцитов и эндотелиальных клеток, передаче сигналов. ICAM-1 - молекула межклеточной адгезии-1. ФВ - фактор Виллебранда. Эндотелин-1. ERK - внеклеточная сигнал-регулируемая киназа, сигнальный путь. ONOO– пероксинитрит, активная форма кислорода. OB-R - рецептор лептина. NF-каппа B - ядерный фактор-каппа B. Универсальный фактор транскрипции, контролирующий экспрессию генов иммунного ответа, апоптоза и клеточного цикла. IL-1 бета - интерлейкин-1 бета. c-Jun N-терминальная киназа (JNK) – киназы, которые связывают и фосфорилируют белок c-Jun. Играют роль в дифференцировке Т-клеток, регулируют апоптоз. Ингибитор каппа-B-киназы. АР-1 активирующий белок-1. NADPH – оксидаза. СОД – супероксиддисмутаза. iNOS - индуцируемая NO-синтаза.